
Welcome to the next chapter in the Data Maker series as we’ll discuss Next Generation Sequencing and the massive impact it has and will continue to have in the research world. Before we dive in, let’s go over a little history. As you can probably imagine by the title “Next Generation”, there was a previous method in place to sequence the human genome. This method was called Sanger Sequencing, invented in the 1970s by British biochemist Frederick Sanger. But since we are all friends here, we’ll call him Freddy.

So what Freddy came up with is something pretty cool (it also won him a couple Nobel prizes :-). He invented a method of copying single-stranded DNA with chemically altered bases. This resulted in short fragments that terminated selectively at an A, C, G, or T (the four nucleotides that make up DNA). Freddy then put the strands together like jigsaw pieces to determine the full sequence. For a cool animation of what this all looks like, check this out:
Needless to say (and no offense to Freddy) is that this is a very time consuming process and doesn’t really scale. It did, however, pave the way for what we now call Next Generation Sequencing. With NGS we can screen more samples and detect multiple variants across target areas of the genome. There are a number of companies that offer multiple methods of sequencing under the NGS umbrella: Pacific BioSciences, Oxford Nanopore and Illumina to name a few. For our purposes, we will focus on Illumina which can be considered the 800 lbs gorilla in the room when it comes to NGS technologies.
Now the idea around NGS is to sequence millions of small fragments of DNA from an entire genome in a single run. The DNA sample is first randomly segmented into smaller sections by a fragmentation process. The sections are then bound to a flow cell and amplified into DNA clusters. They are then sequenced using Illumina’s sequencing by synthesis technology. In this process, the addition of fluorescently labeled nucleotides shows one of four colors, each color of course corresponding to an A, C, G or T. These are detected by laser “excitation” and high-resolution cameras in every run.
After few hundred runs, the instrument will have recorded the sequence of bases in each cluster. The newly identified strings of bases are then assembled into a new sequence (referred to as “de novo”) or aligned to a reference genome.
…and this where the fun begins 🙂
You see, each of those small sections of DNA fragments are kind of useless unless they are “aligned” when you start to look for variants. So what you start out with is something that looks like this:

…err, well, not really this, but you get the concept. It actually looks like this:
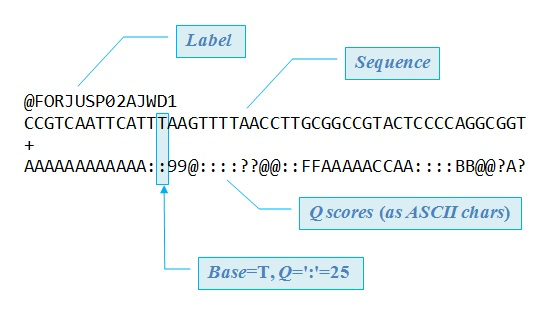
This is a FASTQ file, which is basically a set of unaligned reads. You can actually see the As, C, Gs, and Ts listed along with the encoded quality scores.
The first step here is to look at the Quality Control score to determine if the data even worth moving forward with. You can use a tool like FASTQC to determine this, something in the 30s is good 🙂
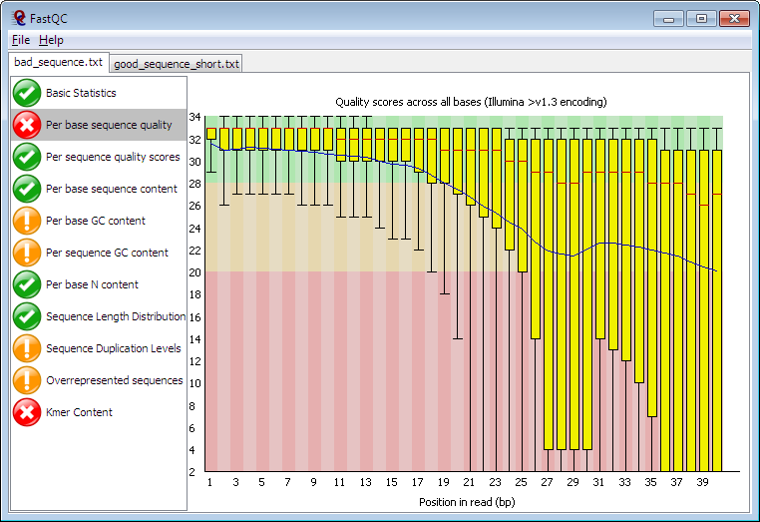
Next up is the alignment phase. This is where we “align” what we just sequenced to a reference genome. As you may know, this can be very compute intensive process. Sample alignment tools / applications are BWA and Bowtie2. The file types you will see after alignment are BAM (Binary Alignment Map) or SAM (Sequence Alignment Map).
![]()

Next up is the variant calling phase, which is also compute intensive. Some of the tools you may have heard of during this phase are Annovar, SAMTools and the Broad Institute’s GATK (amongst many others). The typical output file you will see here is a VCF file or Variant Call Format. This is the smallest file size when compared to BAM and FASTQ and also the most important to the scientist as it shows a list of mutations called SNPs (Single Nucleotide Polymorphisms) that may be relevant to their research.
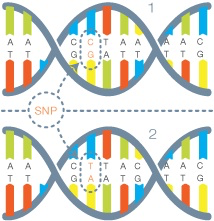
From this point, additional downstream analysis can be done depending on the focus and scope of the project.
Now I would be remiss if I didn’t point out that NGS is used in a variety of ways and there are also different types of sequencing that can be done, Whole Genome, Exome, Methylation..etc. That said and depending on whether or not you are a research hospital, genome center or research institute has a lot to do with what your overall needs would be.
NGS data is also growing, rapidly. A recent Nature article predicts NGS data will outpace both Twitter and YouTube by 2025. Fortunately with the work we’ve been doing at Dell EMC, we are prepared handle the influx of NGS data and help better enable researchers. There is the soup to nuts approach with the Dell EMC HPC System for Genomics, as well as our Emerging Technology Solutions for Life Sciences, and the ultra-cool new solution with Edico Genome’s Dragen Cards, for more on that announcement click here.
I hope you enjoyed this overview of NGS…. and if you don’t remember anything I said, just try to remember this:

Sources:
http://web.uri.edu/gsc/next-generation-sequencing/
https://www.dnalc.org/view/15479-Sanger-method-of-DNA-sequencing-3D-animation-with-narration.html